For any questions or suggestions please contact us
GoTo ...
Guidance
FastML
M1CR0B1AL1Z3R
Selecton
ConSurf
|
Selecton Overview
Introduction
Detecting biologically significant amino-acid sites is important for
such areas as drug design and study of protein function and structure. Conserved sites are indicative of active sites or protein-protein interaction epitopes while highly variable sites may represent sites subjected to positive Darwinian selection. Such positively selected sites may be interpreted as being a consequence of molecular adaptation, which confers an evolutionary advantage to the organism. The Selecton server enables detecting the selective forces at a single amino-acid site. The ratio of non-synonymous (amino-acid altering) to synonymous (silent) substitutions, known as the Ka/Ks ratio, is used to estimate both positive and purifying selection at each amino-acid site.
Input
The minimal input to the server consists of:
- Either a non-aligned file
containing homologous coding DNA sequences, or a codon-aligned file of coding DNA
sequences. The sequences may be in fasta format only. If you are
working with other sequence file formats such as Clustal, Phylip, etc., we suggest
using software such as Bioedit to
convert your format to fasta.
The sequences must be of coding regions only (i.e. composed of triplets of codons) and may not contain internal stop-codons.
A minimum of 3 sequences is required, yet it is recommended to work with
at least 10 sequences (more information on the number of sequences).
-
A query sequence. This is the name of the sequence on which the
results will be displayed. Please copy the name exactly as it appears in
the input sequence file. If Selecton does not find the query name you
specified, the first sequence from the input sequence file will be used
as the query.
Advanced Options
- PDB 3D-structure
Three-dimensional structures of biological macromolecules are available
in the Protein Data Bank (PDB). If a PDB file is available for the
protein you are studying, we highly recommend using it with
Selecton. This will allow you to view the evolutionary selection forces operating on
the protein mapped onto its 3-dimensional structure. In
order to do so, please submit a PDB ID, as well as a chain identifier
under the Advanced options section
(if the molecule has only a single, unnamed chain, specify "none").
- Evolutionary Model
Selecton implements an assembly of different evolutionary codon
models (Yang et al. 2000, Swanson et al. 2003, Doron-Faigenboim et al. 2006). The essence
of these models is that they all assume a prior distribution over the
Ka/Ks ratios (often denoted as ω values). The distributions are approximated using (by
default) eight discrete categories and the Ka/Ks values are computed by
calculating the expectation of the posterior distribution.
The user may choose between the gamma and adapted beta distribution, and
may further choose the number of categories used for the discrete
approximation. The more categories used, the more exact the
computation. However, this is also time consuming. Currently Selecton
allows using between 4 and 14 categories.
Evolutionary models allow performing model testing. Due to a problem of a high false positive rate of
positive selection, it is common to perform extensive significance
testing in order to declare that positive selection is detected. This includes a stage of
comparing the likelihood under a model which assumes positive selection,
to a model which does not allow positive selection, and performing a
likelihood ratio test to decide which model better
fits the data studied.
-
Tree phylogeny
This feature enables the users to supply a more accurate tree, if available. Additionally, the user can supply the tree phylogeny and have the server optimize the branch lengths of this phylogeny.
By default, Selecton constructs a
neighbour-joining tree. As input for the neighbor-joining algorithm, pairwise distances are computed
applying the ML criterion under a codon model (Nielsen and Yang 1998) which assumes no selection
(ω = 1 for all sites and κ = 2). Branch lengths are ML
estimates. We use an expectation maximization
(EM) approach to solve the problem of multivariate optimization in
the case of branch lengths (a similar approach is described in detail in
Friedman et al. 2002).
We note that in general, more accurate tree topologies lead to better
estimate of parameters. Yet, it was previously shown that the detection
of positive selection is in general robust to tree topology inaccuracies
(Pie 2006). Hence, the strategy we adopted in Selecton was to avoid the computationally intense search for the precise tree topology, yet to allow the user to provide a pre-computed phylogenetic-tree as an additional input.
- Genetic code
Eleven different genetic codes are implemented in Selecton, including four different nuclear code variants and seven different mitochondrial code variants. This allows the analysis of genes from organisms and organelles which use non-standard genetic codes.
Methodology
What are Ka/Ks values
The main idea behind the Ka/Ks value is to contrast two types of substitutions events. A substitution is defined synonymous if it does not change the amino-acid sequence. Otherwise it is nonsynonymous. A change in an amino acid due to a nonsynonymous nucleotide change is called a replacement:
Synonymous substitution: CTT (Leucine) → CTA (Leucine)
Non-synonymous substitution: CTT (Leucine) → GTT (Valine)
Thus, the aim of Selecton is to calculate the synonymous rate (Ks) and the non-synonymous rate (Ka), at each codon site.
These rates are normalized, so that in the case of no selection,
Ka/Ks = 1. In the case of purifying selection, Ka decreases, and thus,
Ka/Ks < 1 is indicative of purifying selection. In rare cases where the
opposite pattern is observed (Ka/Ks > 1), "positive Darwinian selection"
is usually invoked as a possible explanation. The idea is that in such
positively selected sites, rapid replacement of an amino acid is advantageous to the organism; hence, mutations are fixed at a rate higher than that of neutral mutations (i.e., the synonymous ones).
Evolutionary models
In essence, an evolutionary model describes in probabilistic terms how characters (in our case - codons) evolve along a
phylogenetic tree. The main goal when building an evolutionary model is
to ensure that it is expressive enough to describe the biological
reality, yet does not over fit the observed sequences. Selecton
implements several codon models, each of which assumes different
biological assumptions. One of the main advantage of these models is that they
enable contrasting different hypotheses, by testing which model better
fits the data at hand. In the case of Selecton, the hypothesis at hand
is whether positive selection is operating on the protein studied, and
this is contrasted with a null hypothesis which assumes there is no
positive selection.
We will give a brief description here of each of the models. All the models in Selecton are
Bayesian models which assume a statistical distribution to account for
heterogenous Ka/Ks values among sites. Since working with continuous
distributions is computtionally intense, the distribution is
approximated with (by default 8) discrete categories. Full details on each of the
models are available in the reference:
- M8 (Yang et al. 2000)
This is the default model for all Selecton runs. It allows for positive
selection operating on the protein. A proportion p0 of
the sites are drawn from a beta distribution (which is defined in the
interval [0,1]), and a proportion
p1(=1-p0) of the sites are drawn from an additional category
ωs (which is constrained to be ≥ 1). Thus, sites drawn
from the beta distribution are sites experiencing purifying selection,
whereas sites drawn from the the ωs category are sites
experiencing either neutral or positive selection. Both p0 and ωs are
estimated using ML.
- M8a (Swanson et al. 2003)
This model is in essence similar to the M8 model, expcept for the fact
that it does not allow for positive selection by setting
ωs = 1. Thus, only neutral and purifying selection are
allowed here. The M8a model is nested in the M8 model: if under the M8
model ωs is estimated as being 1, we obtain the M8a
model. This allows for
hypothesis testing by performing a likelihood
ratio test between the two models to see which model fits the data
better.
- M7 (Yang et al. 2000)
Again, this model is similar to M8, except for the fact that it assumes
only a beta distribution with no additional category. Thus it allows
mainly for purifying selection in the protein. The M7 model is nested in
the M8 model: if p0 is estimated as being 1, the M8 model
collapses to the M7 model.
- M5 (Yang et al. 2000)
This model assumes Ka/Ks among sites are gamma distributed. As opposed
to the beta distribution, the gamma distribution is not constrained, and
thus may allow for purifying, neutral, and positive selection. However,
the disadvantage of the model is that under a discrete categorization of
the distribution it is possible to obtain no category of positive
selection, although positive selection is operating on the protein.
- MEC (Doron-Faigenboim and Pupko 2006)
The MEC model is essentially different from all the above described
models, in that it is the only model in Selecton which takes into account
the differences between different amino-acid replacement
probabilities. This model expands a 20 by 20 amino-acid
replacement probability matrix (such as the commonly used JTT matrix)
into a 61 by 61 sense-codon probability matrix. The rate of transition
(tr) and the rate of transversion (tv) are parameters integrated into the matrix. To take into account the
different selection forces and their intensities, all non-synonymous substitutions in the
matrix are multiplied by ω (for which a prior distribution of gamma
is assumed). It is important to note here that here, ω does not
stand for positive selection, it only stands for the intensity of
selection (purifying or positive). Ka/Ks may be computed from the values
of ω (see reference for more details).
Since replacement probability matrices inherently assume selection, the
MEC model assumes another parameter (denoted as f) which stands for the
proportion of sites undergoing no selection.
The advantage of the MEC model over the other models presented here is
that by treating different amino-acid replacements differently, Ka is
computed differently: under the MEC model, a position with radical
replacements will obtain a higher Ka value than a position with more
moderate replacements.
However, there is no null model nested within the MEC model. Hence, to
perform statistical analyses, Akaike Information Content
(AICc) scores are compared between the MEC and the M8a
models. the lower the AIC score, the the better the fit of the model to
the data, and hence the model is considered more justified.
Significance testing
In order to test whether postive selection is operating on a protein, it
is custom to perform two steps:
- Perform a likelihood ratio test (on the whole gene) between a null model (which doesn't
account for sites under positive selection), and an alternative model
that does (Yang et al. 2000). In brief, this means taking twice the difference between the two likelihood scores of the null
model and the alternative model (which assumes positive selection), and
compare it to a chi-square table.
Selecton enables comparing:
- Between the M8 and M8a models. This comparison assumes DF = 1 (one
additional parameter p0).
- Between the M8 and M7 models. This comparison assumes DF = 2 (two
additional parameters: p0 and ωs).
- Between the MEC model and either the M8a or the M7 model. These
models are not nested and hence the comparison requires comparing AIC
scores.
- Predict whether a site is undergoing positive selection using a
Bayesian approach (i.e. search for sites with an ω which is inferred
to be higher than 1). Selecton infers site-specific Ka/Ks by computing the expectation of the posterior
distribution at each site.
Confidence estimates of the Bayesian estimates
The reliability of the Ka/Ks ratios inferred depends on several factors,
including the reliability of the multiple sequence alignment (MSA), the
number of gaps in the MSA, and the number of homologous sequences
used.In the Bayesian method, a p-value cannot be calculated and thus we
calculate a confidence interval defined by the 5th and 95th percentiles
of the posterior distribution inferred for the position. For positions
with an inferred Ka/Ks > 1, a confidence interval is
estimated. If the lower bound of the confidence interval is larger than
1, the inference of positive selection at this position is considered
reliable. Such positions are colored in dark yellow in the graphical
display of the results.
Under a ML model (relevant only for Selecton version 1)
As opposed to the empirical bayesian model, here a likelihood ratio test
is performed for each site:
The likelihood ratio test (LRT) can be used to compare two nested models: a null model which does not account for sites undergoing positive or purifying selection, and an alternative model that does. The LRT statistic 2d(t) (twice the log likelihood difference) can be approximated by the chi-square distribution where the number of degrees of freedom is equal to the difference in the number of free parameters in the two models. In the selection model the number of free parameters is one (c) whereas in the no-selection model the number of free parameters is zero.
A site is suspected to be under positive selection if the ML estimates of the Ka/Ks ratio of equation (1.5) is greater than one. If the ratio is smaller than one it is suspected to be under purifying selection. To prove that the site is significantly under positive or purifying selection, the LRT is used. Anisimova, Bielawski and Yang (Anisimova et al. 2001) used LRT for detecting adaptive evolution; they found that the use of the chi-square distribution made the LRT conservative. However, the test was powerful when enough lineages were analyzed.
Multiple Testing (relevant only for Selecton version 1)
Setting the significance level at alpha=0.05, which defines the range of the type I error (rejection of the null hypothesis while it is true - false positives) is used for a single statistical test. In our case, multiple tests are involved, since the LRT is applied to each site in the sequence. However, by setting alpha at the same value, the protection against type I error is not efficiently provided.
One known multiple test correction is the Bonferroni correction. The p-Value of each site is multiplied by the number of sites. If the corrected p-Value is still below the error rate alpha, this test will be considered significant. Since the Selecton server outputs the list of p-values at each site, one may multiply each p-value by the number of sites, and verify whether the site is still significantly under positive or purifying selection.
Alternatively, a less stringent correction method, named False Discovery Rate (FDR) was introduced by Benjamini and Hochberg (1995). The FDR is the expected proportion of true null hypotheses rejected out of the total number of null hypotheses rejected. This method of correction will be implemented in Selecton in the near future, thus providing adequate protection against the multiple testing problem.
Output
In each run, Selecton produces an output file called "Selecton Job Status Page". This file is automatically updated every 30 seconds, showing messages regarding the different stages of the server activity.
When the calculation finishes, several links appear:
Codon Ka/Ks scores color-coded on the linear sequence
This link is the main link in Selecton, and includes a projection of the
Ka/Ks scores onto the primary seuqence of the protein, using a 7-color
scale. Shades of yellow (colors 1 and 2) indicate a Ka/Ks ratio > 1, and
shades of bordeaux (colors 3 through 7) indicate a Ka/Ks ratio < 1:
|
1 2 3 4 5 6 7
| | Positive selection Purifying selection |
"Codon Ka/Ks Scores (numerical values)"
This link includes the actual Ka/Ks scores obtained for each codon
position of the target sequence, together with their confidence
estinates (95% confidence interval in the Bayesian model, p-values in the ML model)
"Amino Acids Multiple Sequence Alignment (in Fasta format)"
This links to the amino acid MSA produced by translating the input data
and aligning the translated sequences by
Muscle (Edgar 2004) (this
link is availble when the user supplies a non-aligned sequences file.
"Codons Multiple Sequence Alignment (in Fasta format)"
This links to the codon MSA produced by reverse translation of the amino
acid MSA (or the codon-alignment supplied by the user).
"Phylogenetic Tree"
This links to the textual (Newick) file of the phylogenetic tree
generated during the calculations.
To view a graphic display of the tree, you can use any program for displaying
phylogenies, such as TreeView.
"Likelihood and parameters of Selecton run"
This displays the likelihood of the model which was run given the
data. This value can be further used for statistical
hypothesis testing. Furthermore, this file displays the ML estimates
of all parameters in the model.
"Log-file of Selecton run"
This file contains all the data gathered along the Selecton run. Thus,
it summarizes all the input data, displays the likelihood values as they
converge, and summarizes all the parameters and inferences reached at the end of the run.
"RasMol coloring script source" (available only if a PDB
file was given as input)
This link includes the Chime script commands for
coloring the protein according to the ka/ks grades. This file can
be downloaded and used locally with RasMol
, producing the same color-coded CPK scheme generated by the server.
" View Color Coded Selecton Results"(available only if a PDB
file was given as input)
This link leads to
the graphic visualization of the color-coded
molecule through the
FirstGlance in Jmol interface.
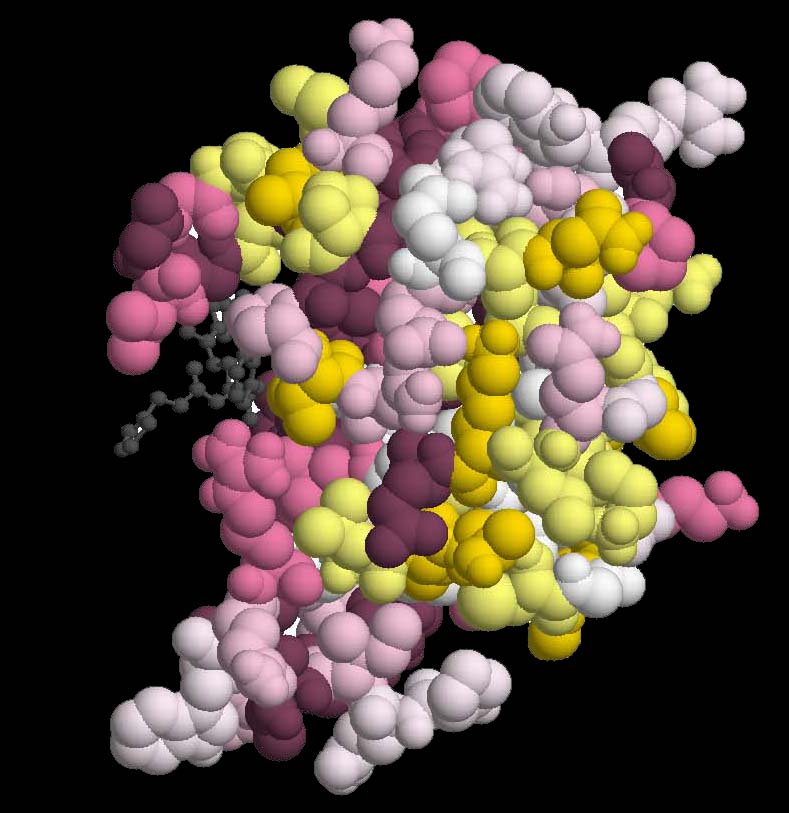
Graphic 3-dimensional Visualization
If A PDB is supplied, the protein is automatically visualized on-line using the FirstGlance in Jmol engine.
The target protein chain is represented as a space-filling model with the conservation grades color-coded onto each amino acid van-der-Waals surface. All other chains in the PDB file are displayed in backbone representation and all ligands are presented in ball-and-stick representation. The user can further control the modes of representation, using the FirstGlance in Jmol visualization capabilities.
References
- Anisimova M, Bielawski JP, Yang Z (2001) Accuracy and power of the likelihood ratio test in detecting adaptive molecular evolution. Mol Biol Evol 18:1585-92.
- Benjamini Y, Hochberg Y (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. Journal of the Royal Statistical Society B 57:289-300
- Doron-Faigenboim, A. and Pupko, T. (2006) A Combined Empirical and Mechanistic Codon Model. Mol Biol Evol, 24, 388-397
- Goldman N, Yang Z (1994) A codon-based model of nucleotide
substitution for protein-coding DNA sequences. Mol Biol Evol
11:725-36
- Edgar, R.C. (2004) MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics 5(1): 113.
- Nielsen, R. and Yang, Z. (1998) Likelihood models for detecting positively selected amino-acid sites and applications to the HIV-1 envelope gene. Genetics, 148, 929-936
- Friedman, N., Ninio, M., Pe'er, I. and Pupko, T. (2002) A structural EM algorithm for phylogenetic inference. J Comput Biol, 9, 331-353.
30.
- Pie, M.R. (2006) The influence of phylogenetic uncertainty on the detection of positive Darwinian selection. Mol Biol Evol, 23, 2274-2278
- Pupko T, Bell RE, Mayrose I, Glaser F, Ben-Tal N (2002) Rate4Site: an algorithmic tool for the identification of functional regions in proteins by surface mapping of evolutionary determinants within their homologues. Bioinformatics 18 Suppl 1:S71-77
- Swanson, W.J., Nielsen, R. and Yang, Q. (2003) Pervasive adaptive evolution in mammalian fertilization proteins. Mol Biol Evol, 20, 18-20
- Yang Z, Nielsen R, Goldman N, Pedersen AM (2000) Codon-substitution
models for heterogeneous selection pressure at amino acid
sites. Genetics 155:431-449
To the top
|